
近期,哈佛大学和麻省理工学院的研究团队在微生物群落研究方法学上取得重要突破,发明了微生物高通量单细胞基因组学技术——Microbe-seq。相关成果以研究长文(Research Article)的形式于6月3日在Science上以High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome为题发表。

Microbe-seq技术集成了多种液滴微流控操作技术和定制开发的生物信息学分析手段,不需要培养即可从复杂微生物群落中获取成千上万个单细胞微生物的基因组信息,并组装出高质量的菌株水平基因组,从而能够在不损失分辨率或广泛物种适用性的基础上探究微生物群落的基因组。该方法应用面广泛,可用于具有复杂微生物群落的样本,如粪便、土壤和海洋等,在微生态研究中具有极大的市场应用潜力。
研究背景:
微生物群落(microbiome)存在于各种不同的生态系统中,典型的微生物群落包括土壤、海洋或江湖等环境微生物以及人体或动物肠道微生物等。其中,人体肠道微生物(human gut microbiome)的组成和功能与人类的健康和疾病息息相关。每个个体的肠道微生物群组成都是特异的,虽然人们经常携带相似的微生物物种,但是不同个体有不同的菌株,不同菌株表现出巨大的基因组差异。菌株之间的这些基因组变异可能导致重要性状的差异,如抗生素耐药性、代谢能力以及与宿主免疫系统的相互作用,这可能对人类健康造成严重后果。
肠道微生物群落中的微生物行为不仅受特定菌株的存在影响,还与它们之间的相互作用密切相关,比如噬菌体调节细菌组成,以及微生物细胞之间遗传物质的转移。为了更好地理解这些微生物的影响,我们需要对特定微生物的基因和功能途径有详细的了解。
然而,要想阐明微生物菌株中的这些信息,可能会面临相当大的挑战。因为现有技术往往仅能在物种水平上对微生物进行分析,难以获知菌株水平上的重要差异。因此,如果能从微生物群落中解析出菌株水平的高质量基因组,那么这将极大地帮助我们理解对肠道微生物行为及其对健康的影响。
现有技术短板:
1、 目前宏基因组测序技术被广泛应用到探索复杂微生物群落,包括人类肠道微生物的组成,然而,宏基因组学通常无法解析菌株水平的基因组。
2、 基于培养的方法只能偏向于容易培养的少数微生物菌株,且成本偏高。
3、 基于微孔板的单细胞测序虽然可以产生菌株水平的基因组,但是成本等限制导致这种方法只能获得有限数量的微生物菌株。
技术和分析原理:
为了解决上述问题,哈佛大学和麻省理工学院的研究团队开发了一种微生物高通量单细胞基因组测序技术Microbe-seq,这一技术的实验原理如下:
1. 利用液滴微流控技术,研究团队将成千上万的微生物单独地包裹在液滴中;
2. 在每个液滴中,裂解微生物并释放DNA;
3. 进行全基因组扩增,将扩增出来的DNA打断并连上接头,随后在液滴中用带有特定序列的标签标记液滴内DNA;
4. 将所有液滴内的DNA合并,进行建库测序。
由于液滴内DNA标记的标签对于每一个液滴都是不同的,因而将测序后的序列按照DNA标签进行区分,即可得到每一个液滴内单个微生物的基因组信息。这些具有相同barcode的测序序列的集合即称为一个single amplified genome (SAG)。
对于复杂微生物群落,其中大多数微生物的参考基因组一般是未知的。而单个SAG涵盖的基因组信息一般在全基因组的50%以内,无法直接被用于参考基因组。这为微生物组的单细胞基因组分析带来了一个独特的难题,为了得到高质量的微生物群落的参考基因组,研究团队还发展了一套通用的生信分析流程:
1. 通过提取并比对各单细胞的基因组标志性信息(genomic signature),将样品中来自同一物种的单细胞微生物识别出来,然后合并在一起来组装出物种水平的参考基因组;
2. 进一步将单个微生物的基因组和参考基因组对比,识别来自于不同菌株的单细胞微生物并进行基因组组装。
通过这种方法,研究人员获得了样品中多种不同物种及物种中不同菌株的基因组。
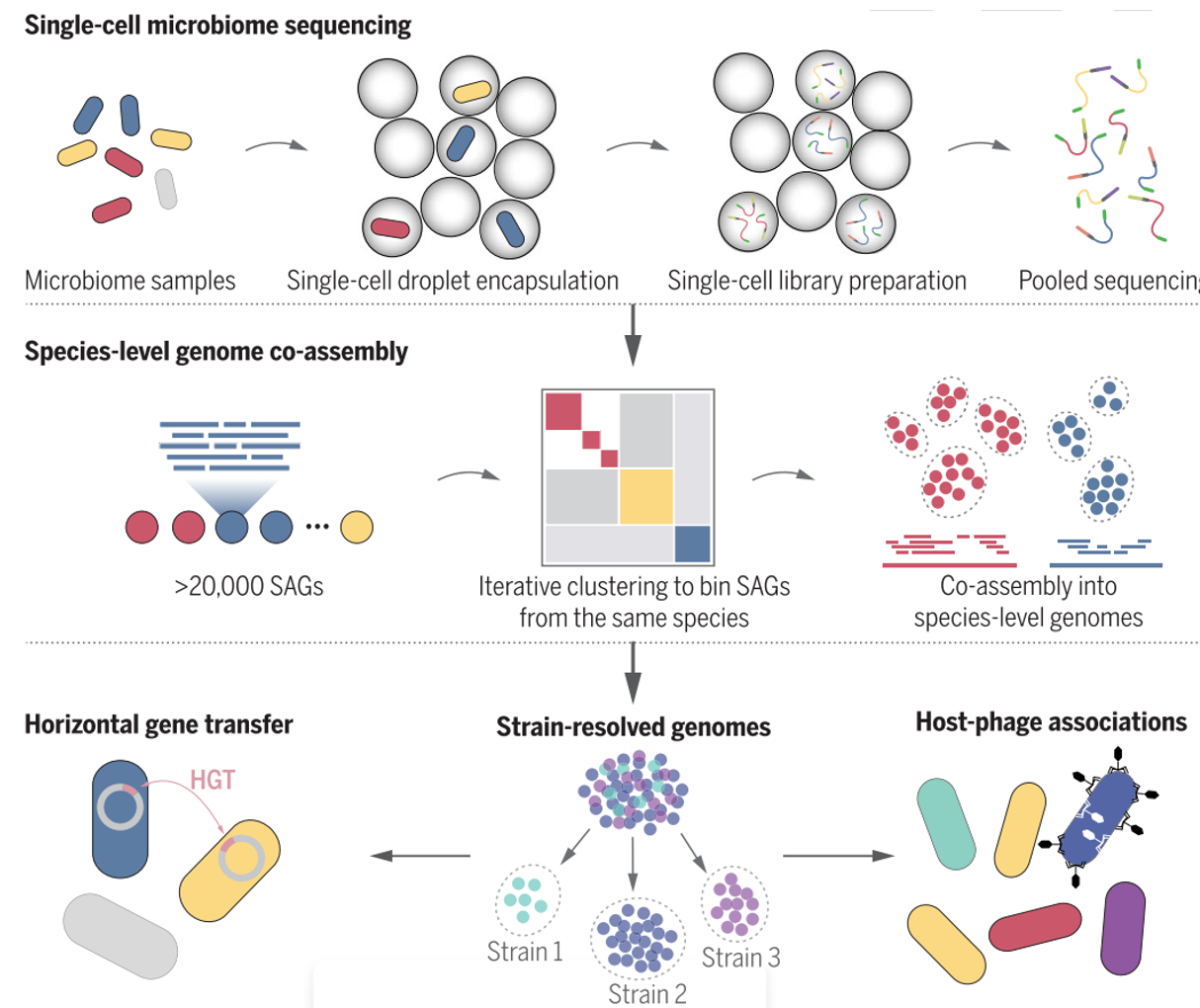
Microbe-seq概览
Microbe-seq性能评估——基于模拟微生物群落样本:
为了表征每一个SAG所包含的基因组信息,研究人员用四种已知菌株构建了模拟微生物群落样本并应用Microbe-seq方法对其进行测试。这四种菌株包含了两种实验室常见的革兰氏阴性菌(大肠杆菌、肺炎克雷伯菌)和两种革兰氏阳性菌(枯草芽孢杆菌、金黄色葡萄球菌),经过实验,共得到了5497个SAG。为了验证单个SAG中的序列是否来自于单个微生物,研究人员用纯度来表征每个SAG中单微生物基因组的比例,SAG纯度的定义是一个SAG的所有能够比对到四种已知菌株的测序序列中,比对到四种菌株中的序列比例的最大值(即比对到上述四种菌株任一基因组的序列数/SAG比对到所有四种菌株的基因组总序列数的比值的最高值)。比对结果显示,在模拟样品中,84%(4612)的SAG纯度超过95%,表明绝大多数SAG都来自单一微生物(见下图B)。
研究人员同时也统计了单个SAG包含的基因组信息的比例,对于革兰氏阴性菌大肠杆菌和肺炎克雷伯菌,平均基因组覆盖率分别为8%和10%;而对于革兰氏阳性菌枯草芽孢杆菌和金黄色葡萄球菌,比例为17%和25%。为了获取完整的基因组信息,一种常用方法是合并来自相同物种的多个SAG的基因组信息。测试发现,对于上述四种菌株,仅需要几十个SAG就能覆盖基本完整的基因组(见下图C)。
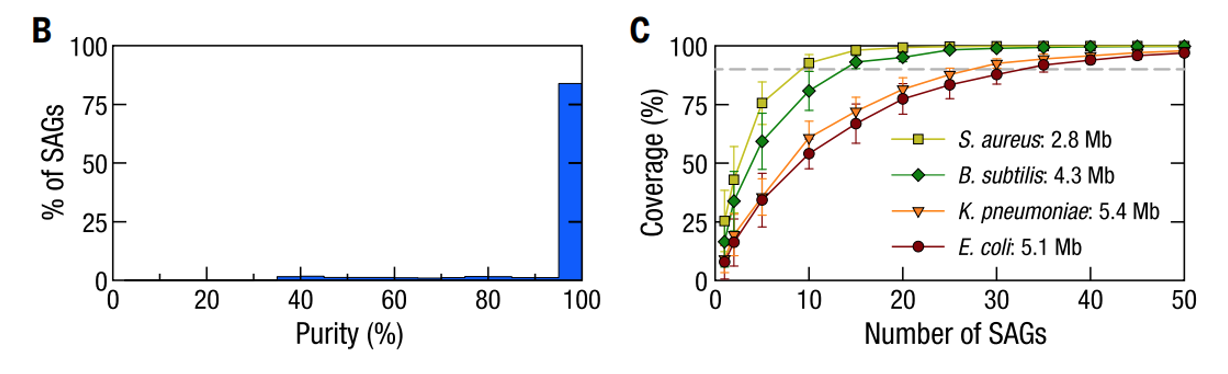
基于模拟群落样本评估Microbe-seq性能
Microbe-seq应用——基于复杂的人体肠道群落样本:
1、 物种水平的高质量基因组组装:
研究人员将Microbe-seq方法应用于一个健康人捐赠的7份粪便样本,对应样品的宏基因组测序结果和分离培养鉴定结果在之前已经被报道,这些已经报道的工作为多维度评估Microbe-seq的结果提供了可能。通过Microbe-seq,研究人员从每个样本中得到了1000-7000个SAG,总数为21914个,每个SAG平均包含约70000条序列。
为了更好地对这些SAG进行组装,研究人员开发了一种不参考外部基因组的方法,将同一物种多个相似SAG进行共组装,然后将平均核苷酸同一性(ANI)值超过95%的基因组归为同一物种,并将它们的序列进行合并,进而获得物种水平的基因组。
组装获得的微生物基因组质量,一般用完整度和污染度两个指标进行评估:完整度>95%且污染度<5%的基因组为高质量,完整度>50%且污染度<10%为中等质量,其余则为低质量。在这个健康人的肠道微生物中,通过Microbe-seq发现了76个物种,其中52个物种具有高质量基因组,24个物种具有中等质量基因组(见下图)。超过四分之三(16723)的SAG属于这76个物种中的一个,表明Microbe-seq得到的大部分SAG的参考基因组已成功重建。
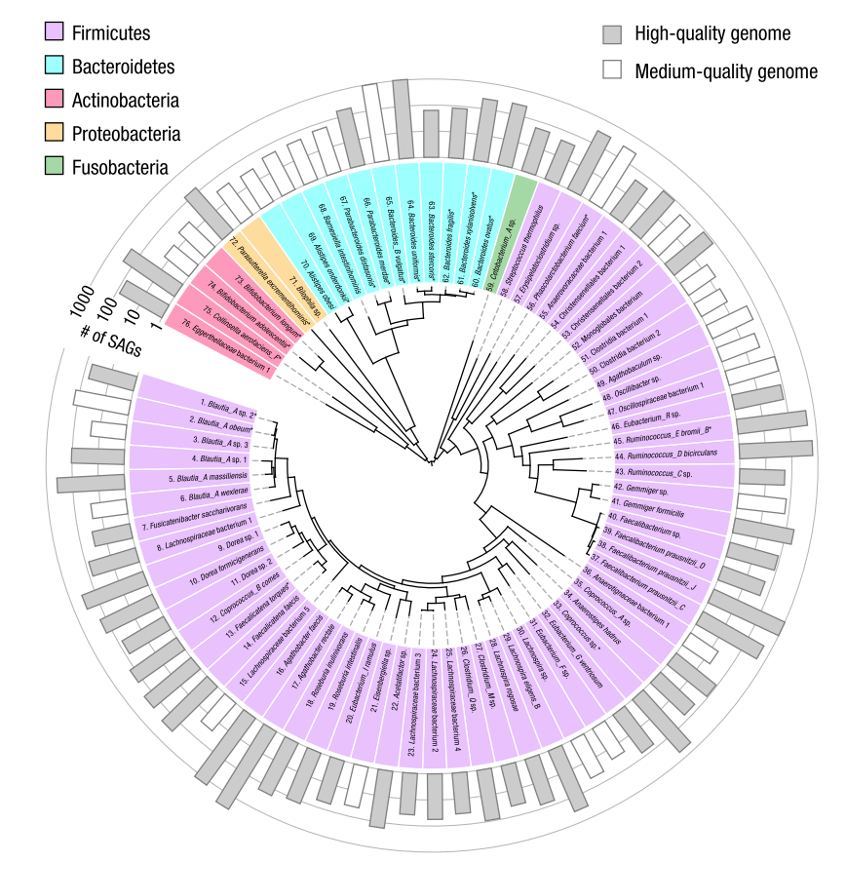
个体肠道微生物群中76种细菌的共组装基因组
注:柱子长度代表在其共组装中使用的SAG数目,灰色柱子代表基因组组装质量高的物种。
2、 可获得媲美“金标准”水准的可靠基因组:
将共组装基因组与可分离培养细菌的“金标准”基因组进行比较,结果发现76个物种中,有19个物种已经被成功分离培养。在这19个物种中,有17个物种的共组装基因组与“金标准”基因组高度一致,ANI超过99.5%,表明Microbe-seq可以获得与分离培养相似水平的可靠基因组。
3、 可获得与宏基因组一致的微生物多样性:
通过与宏基因组微生物多样性结果对比,发现通过Microbe-seq检测到的属同宏基因组结果重合度高达96.9%-99.8%。具体到每个属的相对丰度,两种技术有一些不同,但是同一个门内的微生物的丰度趋势在两种方法中一致(见下图),表明Microbe-seq与宏基因组获得的微生物多样性非常类似。
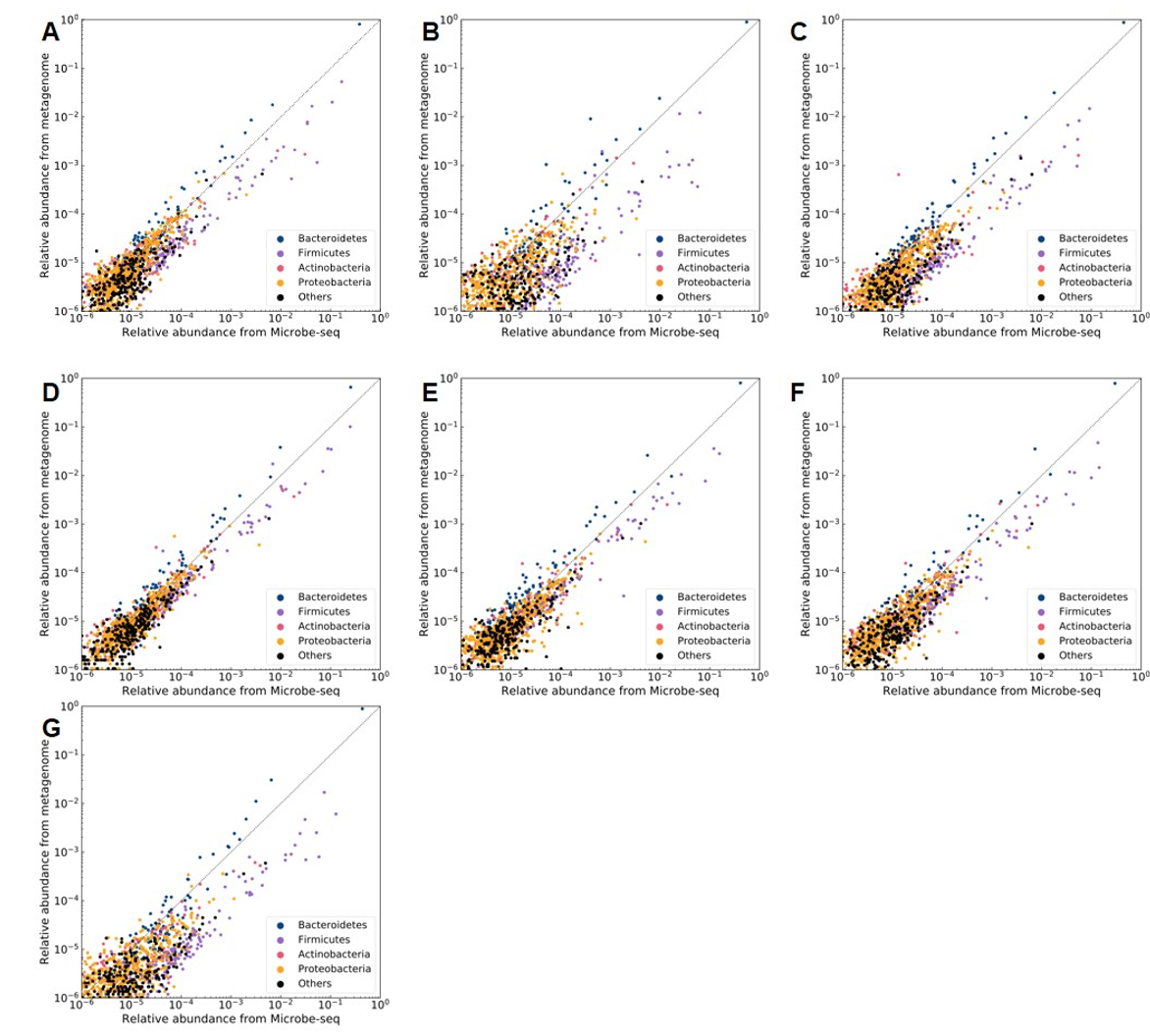
Microbe-seq与宏基因组微生物多样性结果对比(A-G分别展示对于7个样品的结果)
4、 可精确解析菌株水平基因组,同时发现未培养的全新菌株:
将SAG重新比对到物种水平的组装基因组,可以得到SAG相较于基因组的SNP信息。依照SNP信息对普通似杆菌(Bacteroides vulgatus)的SAG进行层次聚类(hierarchical clustering)并进行UMAP降维可视化,SAG分为四个明显分离的聚类簇(见下图A);每个普通似杆菌的SAG聚类簇具有一致的基因型,而与其它SAG簇的基因型重叠程度很低(见下图B),这说明每个SAG聚类簇代表不同的菌株。
通过共组装,获得了菌株A和B的高质量基因组、菌株C的中等质量基因组以及菌株D的低质量基因组,其中,菌株A和菌株C与分离培养的两个菌株的基因组一致,菌株B为Microbe-seq所鉴定到的全新菌株(见下图C),这些结果共同证明了Microbe-seq能够正确鉴定到已知菌株和尚未培养的潜在新菌株,同时实现其基因组的精确共组装。
利用这种方法,在该个体的76个微生物物种中,总计发现了10个具有多个不同的菌株的物种,并且组装了对应的菌株水平的基因组。在前文中提到的19个已经被成功分离培养的物种中,有两个物种的共组装基因组与“金标准”基因组具有一定偏差,ANI小于99.5%。而这两种物种都具有多个菌株,解释了共组装基因组与“金标准”基因组偏差存在的原因。
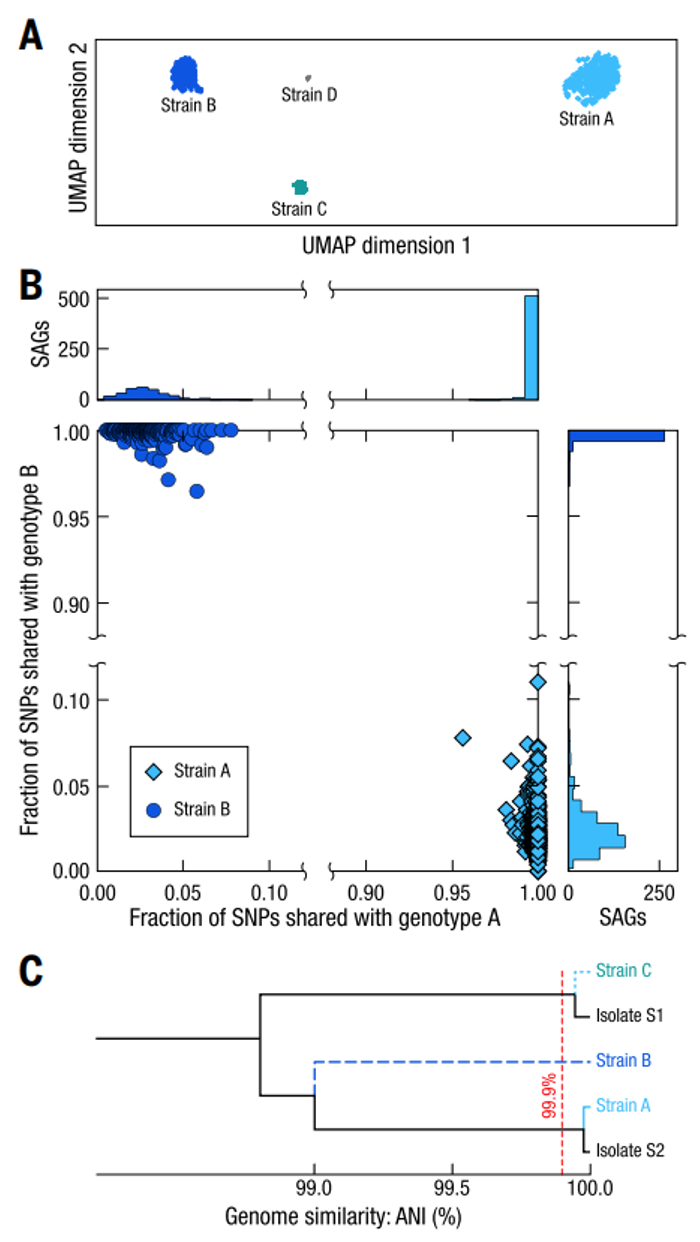
人类肠道微生物群中普通拟杆菌的菌株水平基因组解析
5、可在菌株水平分析水平基因转移事件:
除此之外,该团队利用Microbe-seq构建了单个个体肠道微生物群中菌株的水平基因转移(HGT)网络,并且发现同一细菌门中的菌株之间的转移明显大于不同细菌门中的菌株之间的转移(见下图)。
这些数据表明Microbe-seq能够在多个菌株中广泛而有力地检测HGT,从而为研究菌群中多种微生物的相互作用提供了新的工具。
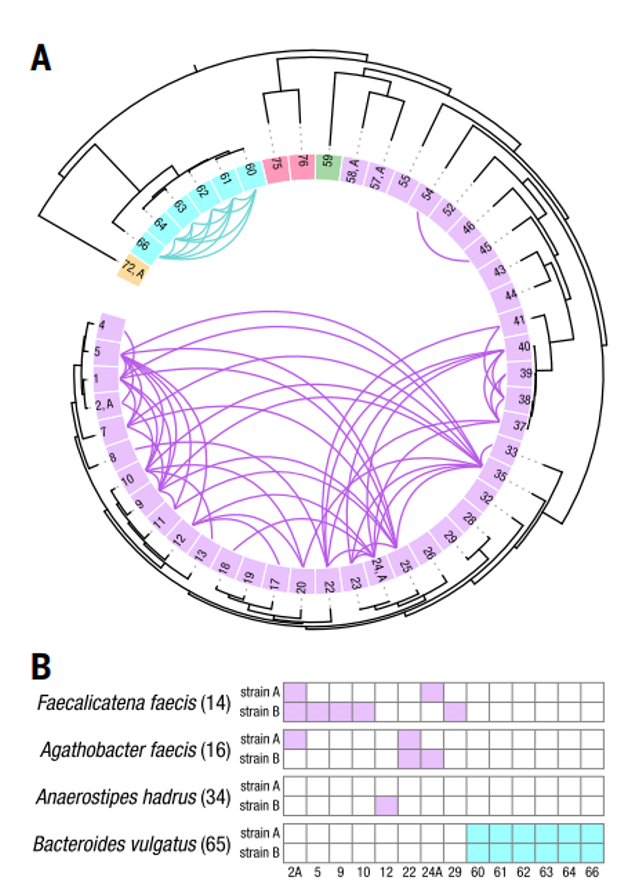
在细菌菌株之间进行水平基因转移(HGT)分析
6、可在菌株水平分析宿主-噬菌体关联信息:
在Microbe-seq的实验过程中,每个液滴不仅包裹了单个细菌,还包裹了液滴中与之共生的噬菌体,为探索宿主-噬菌体关联提供了直接手段。本研究不但发现普通似杆菌是该个体中噬菌体crAssphage的体内宿主物种,更精确地在菌株水平发现普通似杆菌菌株A与噬菌体crAssphage存在明显的关联。
这些数据表明,Microbe-seq具有独特的优势,不仅可以为单个物种,而且可以更精确地为特定菌株建立准确的体内宿主噬菌体关联。
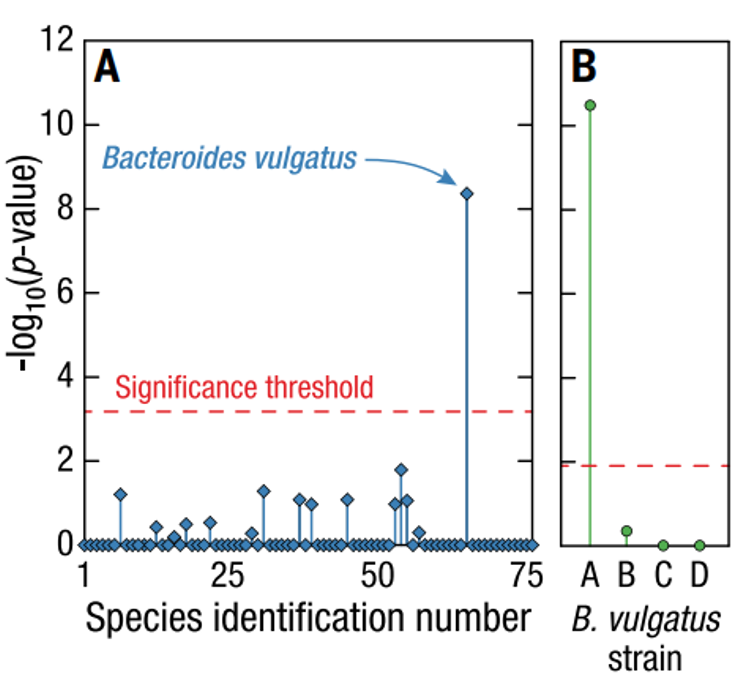
宿主噬菌体与菌株特异性的关联分析
研究总结:
Microbe-seq技术集成了多种液滴微流控操作技术和定制开发的生物信息学分析手段,不需要培养即可从复杂微生物群落中获取成千上万个单细胞微生物的基因组信息,并组装出高质量的菌株水平基因组(包含大量尚未被培养的菌株),从而能够在不损失分辨率或广泛物种适用性的基础上探究微生物群落的基因组。同时,该技术还可精确解析菌株水平基因组、发现未培养的潜在的新菌株;进而可在菌株水平对HGT、宿主-噬菌体关联、功能基因和代谢通路进行深入研究。
该方法可同时适用于其它复杂的微生物群落,如土壤和海洋中的微生物群落,有望成为医学和环境微生态研究的主流技术,对于发掘有潜在应用价值的微生物资源、探究种间/种内关系具有重要的理论和现实意义。
该文章的共同第一作者为哈佛大学化学和化学生物学系郑文山博士(现为墨卓生物CTO)和麻省理工学院生物工程系赵诗杰博士,共同通讯作者为哈佛大学工程与应用科学学院的David Weitz教授(墨卓生物联合创始人)、Peter J. Lu(陆述义)博士和麻省理工学院生物工程系的Eric J. Alm教授。
原文链接:http://doi.org/10.1126/science.abm1483

尊敬的 先生/女士
您已注册成功,注册信息及注意事项已发到联系人及参会人邮箱,请注意查收。如未收到,请联系大会联系人。

